561276
NORMAS LEGALES
Sábado 12 de setiembre de 2015 /
El Peruano
Artículo 4. La presente Resolución Ministerial no
libera ni exonera del pago de impuestos o de derechos
aduaneros cualquiera sea su clase o denominación.
Regístrese, comuníquese y publíquese.
ANA MARÍA SÁNCHEZ DE RÍOS
Ministra de Relaciones Exteriores
1286847-1
RESOLUCIÓN MINISTERIAL
Nº 805/2015-RE
Lima, 11 de setiembre de 2015
CONSIDERANDO:
Que, en la Declaración Conjunta suscrita en el marco
de la Visita de Estado a Colombia del Presidente de la
República, señor Ollanta Moisés Humala Tasso, realizada
el 11 de febrero de 2014, se dispuso convocar a la IV
Reunión del Mecanismo de Consulta y Coordinación
Política 2+2, en fecha próxima a acordar por los canales
diplomáticos;
Que, en la reunión de trabajo que tuvo lugar en
la ciudad de Lima el 25 de mayo del presente año, las
Cancilleres del Perú y Colombia acordaron llevar a cabo
la Primera Reunión de la Comisión Binacional de la Zona
de Integración Fronteriza (CBZIF), a nivel de Cancilleres,
en el contexto de las reuniones de alto nivel en Seguridad
y Defensa y Desarrollo Fronterizo;
Que, el 14 de setiembre de 2015, se realizarán
reuniones bilaterales para la preparación de los
documentos y exposiciones que la Secretaría Ejecutiva
presentará en la Primera Reunión de la Comisión
Binacional para la zona de integración fronteriza con
Colombia, el informe escrito y exposición sobre los
avances de la ejecución del plan, así como elaboración
de la propuesta y exposición de la estrategia para el
fortalecimiento de la ejecución del plan;
Que, el Gobierno de Colombia propuso realizar la
IV Reunión del Mecanismo de Consulta y Coordinación
Política (2+2), el 15 de setiembre de 2015, en la ciudad de
Bogotá, D.C., República de Colombia;
Que, en la misma fecha, las Ministras de Relaciones
Exteriores del Perú y Colombia inaugurarán la Primera
Reunión de la Comisión Binacional de la Zona de
Integración Fronteriza (CBZIF);
Que, es necesario que el Director de Desarrollo
e Integración Fronteriza, de la Dirección General de
América, asista a las reuniones antes mencionadas a
fin de asegurar un seguimiento diplomático y político
adecuado de la posición del Perú en el marco de ese
mecanismo;
Teniendo en cuenta la Hoja de Trámite (GAC) N.°
4658, del Despacho Viceministerial, de 10 de setiembre
de 2015; y los Memorandos (DGA) N.° DGA0777/2015,
de la Dirección General de América, de 1 de setiembre
de 2015; y (OPR) N.° OPR0282/2015, de la Oficina de
Programación y Presupuesto, de 10 de setiembre de
2015, que otorga certificación de crédito presupuestario
al presente viaje;
De conformidad con la Ley N.º 27619, Ley que regula
la autorización de viajes al exterior de servidores y
funcionarios públicos, modificada por la Ley N.º 28807, y
su Reglamento aprobado por Decreto Supremo N.º 047-
2002-PCM y sus modificatorias, la Ley N.º 28091, Ley
del Servicio Diplomático de la República, su Reglamento
aprobado por Decreto Supremo N.º 130-2003-RE y
sus modificatorias; el Reglamento de Organización
y Funciones del Ministerio de Relaciones Exteriores,
aprobado por Decreto Supremo N.° 135-2010-RE; y el
numeral 10.1 del artículo 10 de la Ley N.º 30281, Ley de
Presupuesto del Sector Público para el Año Fiscal 2015;
SE RESUELVE:
Artículo 1. Autorizar el viaje, en comisión de
servicios, del Ministro en el Servicio Diplomático de
la República Luis Rodomiro Hernández Ortiz, Director
de Desarrollo e Integración Fronteriza, de la Dirección
General de América, a la ciudad de Bogotá, D.C.,
República de Colombia, del 14 al 15 de setiembre
de 2015, por las razones expuestas en la parte
considerativa de la presente resolución.
Artículo 2. Los gastos que irrogue el cumplimiento
de la presente comisión de servicios, serán cubiertos
por el pliego presupuestal del Ministerio de Relaciones
Exteriores, Meta 0083906 Conducción y Asesoramiento
de Líneas de Política Exterior e Institucional, debiendo
rendir cuenta documentada en un plazo no mayor de
quince (15) días calendario, al término del referido
viaje, de acuerdo con el siguiente detalle:
Nombres y Apellidos
Pasaje
aéreo clase
económica
US$
Viáticos
por día
US$
N.° de
días
Total
viáticos
US$.
Luis Rodomiro Hernández Ortiz
995,00
370,00
2
740,00
Artículo 3. Dentro de los quince (15) días
calendario, posteriores a su retorno al país, el citado
funcionario diplomático deberán presentar a la Ministra
de Relaciones Exteriores, un informe detallado de las
acciones realizadas y los resultados obtenidos en el
viaje autorizado.
Artículo 4. La presente Resolución Ministerial no
libera ni exonera del pago de impuestos o de derechos
aduaneros cualquiera sea su clase o denominación.
Regístrese, comuníquese y publíquese.
ANA MARÍA SÁNCHEZ DE RÍOS
Ministra de Relaciones Exteriores
1286847-2
SALUD
Modifican Reglamento para el Registro,
Control y Vigilancia Sanitaria de Productos
Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios
dECRETO SUpREMO
N° 029-2015-SA
EL PRESIDENTE DE LA REPÚBLICA
CONSIDERANDO:
Que, la Ley Nº 29459, Ley de los Productos
Farmacéuticos, Dispositivos Médicos y Productos
Sanitarios, define y establece los principios, normas,
criterios y exigencias básicas sobre los productos
farmacéuticos, dispositivos médicos y productos
sanitarios de uso en seres humanos, en concordancia
con la Política Nacional de Salud y la Política Nacional de
Medicamentos;
Que, mediante Decreto Supremo Nº 016-2011-
SA, se aprobó el Reglamento para el Registro, Control
y Vigilancia Sanitaria de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios, modificado
por Decretos Supremos Nºs. 001-2012-SA, 016-2013-SA
y 010-2015-SA;
Que, resulta necesario modificar e incorporar algunos
artículos del citado Reglamento, a efecto de perfeccionar
la aplicación de la Ley Nº 29459, Ley de los Productos
Farmacéuticos, Dispositivos Médicos y Productos
Sanitarios;
Que, en concordancia con los Decretos Leyes Nºs.
25629 y 25909, así como el artículo 1 del Decreto
Supremo N° 149-2005-EF, que dicta disposiciones
reglamentarias al Acuerdo sobre Obstáculos Técnicos
al Comercio en el ámbito de bienes y al Acuerdo

561277
NORMAS LEGALES
Sábado 12 de setiembre de 2015
El Peruano
/
General sobre el Comercio de Servicios, en el ámbito
de servicios, de la Organización Mundial del Comercio,
estipula que los trámites o requisitos que afecten de
alguna manera la libre comercialización interna o la
exportación o importación de bienes o servicios podrán
aprobarse únicamente mediante Decreto Supremo
refrendado por el Ministro de Economía y Finanzas y
por el del Sector involucrado;
De conformidad con lo establecido en el numeral 8
del artículo 118 de la Constitución Política del Perú, el
Decreto Ley Nº 25629, el Decreto Ley Nº 25909 y la Ley
Nº 29158, Ley Orgánica del Poder Ejecutivo;
DECRETA:
Artículo 1.- Modificación del Reglamento para el
Registro, Control y Vigilancia Sanitaria de Productos
Farmacéuticos, Dispositivos Médicos y Productos
Sanitarios
Modifíquense los artículos 6, 9, 17, 40, 100, 122,
123, 138, primer y tercer párrafo del artículo 174 del
Reglamento para el Registro, Control y Vigilancia Sanitaria
de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios, aprobado por Decreto Supremo Nº
016-2011-SA, de acuerdo al siguiente detalle:
"Artículo 6.- De la circulación de productos o
dispositivos con características no autorizadas
No podrán circular en el mercado productos o
dispositivos con características diferentes a las autorizadas
en el registro sanitario o certificado de registro sanitario.
Todas las modificaciones o cambios posteriores a
lo declarado para la obtención del registro sanitario y
certificados de registro sanitario, según corresponda,
deben ser previamente comunicados o en su caso
solicitados a la Autoridad Nacional de Productos
Farmacéuticos, Dispositivos Médicos y Productos
Sanitarios, en la forma y condiciones que establece el
presente Reglamento."
"Artículo 9.- Países de alta vigilancia sanitaria
Para efectos de lo señalado en la Ley y el presente
Reglamento, se consideran como países de alta vigilancia
sanitaria los siguientes: Francia, Holanda, Reino Unido,
Estados Unidos de América, Canadá, Japón, Suiza,
Alemania, España, Australia, Dinamarca, Italia, Noruega,
Bélgica, Suecia, la República de Corea y Portugal."
"Artículo 17.- Adhesiones y añadiduras al rotulado
del producto o dispositivo
La información de los rotulados de los productos o
dispositivos a que se refiere el presente Reglamento
debe expresarse en idioma español, con impresiones
de caracteres indelebles, fácilmente legibles y visibles.
Adicionalmente pueden presentarse en otros idiomas,
siempre que dicha información corresponda a la que obra
en el registro sanitario o notificación sanitaria obligatoria
del producto o dispositivo.
El rotulado de los productos o dispositivos, no puede
consignar más información que la aprobada al otorgarse
el registro sanitario, salvo excepciones que considere la
Autoridad de Salud. No se considera modificación del
rotulado la incorporación de tal información.
En los rotulados no se pueden adherir etiquetas para
corregir o agregar información, salvo las que tengan
por objeto señalar nombre, dirección, registro único del
contribuyente del importador y el nombre del director
técnico o cualquier otra información por indicación expresa
de la Autoridad Nacional de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios.
Cuando, por razones de seguridad, la Autoridad
Nacional de Productos Farmacéuticos, Dispositivos
Médicos y Productos Sanitarios, de oficio, disponga alguna
modificación al rotulado autorizado de los productos o
dispositivos, dicha modificación debe ser agregada en
impresión de carácter indeleble.
Para el caso de productos o dispositivos
terminados nacionales e importados, se permite el
reacondicionamiento del envase mediato o inmediato,
cambio de inserto o manual de instrucciones a efectos
que pueda contar con la información autorizada en el
registro sanitario o notificación sanitaria obligatoria. Para
el reacondicionamiento, debe solicitarse autorización
a la Autoridad Nacional de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios, conforme a
lo estipulado en el Reglamento correspondiente.
El reacondicionamiento en productos terminados
consiste en colocar al mismo en un nuevo envase
mediato o secundario, inclusión o cambio de inserto o
agregar información en el envase mediato o inmediato
cuya impresión debe ser clara, legible e indeleble.
Para el caso de dispositivos médicos se permite el
reacondicionamiento agregando información en el
envase mediato o inmediato cuya impresión debe
ser clara, legible e indeleble, cambio de inserto o
manual de instrucciones. En el caso de productos o
dispositivos terminados, el rotulado mediato debe
consignar el nombre del Laboratorio que realiza el
reacondicionamiento.
El rotulado no puede estar impreso ni adherido en la
superficie interna de los envases mediatos e inmediatos,
a excepción de los envases mediatos de la unidad de
venta mínima en envases dispensadores."
"Artículo 40.- Requisitos para la inscripción y
reinscripción de especialidades farmacéuticas
A.Categoría 1: Inscripción y reinscripción de
especialidades farmacéuticas cuyos Ingrediente(s)
Farmacéutico(s) Activo(s) IFA(s) o asociaciones
se encuentran en el Petitorio Nacional Único de
Medicamentos Esenciales:
(...)
Para la presentación de solicitudes de reinscripción
de las especialidades farmacéuticas que hayan
obtenido registro sanitario al amparo de la Ley N°
29316, incluyendo el requisito de estudios o información
técnica de seguridad y eficacia correspondiente y
cuya información de su registro sanitario se encuentre
actualizada, el titular del registro sanitario quedará
exceptuado de presentar los requisitos señalados en
los numerales 2, 3, 4, 9 y el inserto señalado en el
numeral 8 del presente literal. Asimismo, los estudios
de estabilidad a largo plazo, previstos en el numeral 6
de este artículo, se tendrán por presentados en dicho
procedimiento de reinscripción, por medio de una
declaración jurada que exprese que dichos estudios
no han sufrido variación y, en caso de haber sufrido
cambios, éstos se encuentren autorizados. El desarrollo
del estudio debe estar acorde con lo dispuesto en la
Directiva vigente al momento de solicitar la reinscripción.
Para la presentación de solicitudes de reinscripción
de las especialidades farmacéuticas que hayan
obtenido registro sanitario al amparo de la Ley N°
29459, incluyendo el requisito de sustento de seguridad
y eficacia correspondiente y cuya información de
su registro sanitario se encuentre actualizada, el
titular del registro sanitario quedará exceptuado de
presentar los requisitos señalados en los numerales
2, 3, 4, 9 y el inserto señalado en el numeral 8 del
presente literal. Los estudios de estabilidad a largo
plazo, previstos en el numeral 6 de este artículo, se
tendrán por presentados en dicho procedimiento de
reinscripción, por medio de una declaración jurada
que exprese que dichos estudios no han sufrido
variación y, en caso de haber sufrido cambios, éstos
se encuentren autorizados. El desarrollo del estudio
debe estar acorde con lo dispuesto en la Directiva
vigente al momento de solicitar la reinscripción.
B.Categoría 2: Inscripción o Reinscripción en el
Registro Sanitario de especialidades farmacéuticas
cuyos Ingrediente(s) Farmacéutico(s) Activo(s)
IFA(s) o asociaciones no se encuentren en el Petitorio
Nacional Único de Medicamentos Esenciales y se
encuentran registrados en países de alta vigilancia
sanitaria.
(...)

561278
NORMAS LEGALES
Sábado 12 de setiembre de 2015 /
El Peruano
Para la presentación de solicitudes de reinscripción
de las especialidades farmacéuticas que hayan obtenido
registro sanitario al amparo de la Ley N° 29316, incluyendo
el requisito de estudios o información técnica de
seguridad y eficacia correspondiente y cuya información
de su registro sanitario se encuentre actualizada, el titular
del registro sanitario quedará exceptuado de presentar
los requisitos señalados en los numerales 2, 3, 4, 9 y
el inserto señalado en el numeral 8 del presente literal.
Los estudios de estabilidad a largo plazo, previstos en el
numeral 6 de este artículo, se tendrán por presentados en
dicho procedimiento de reinscripción, por medio de una
declaración jurada que exprese que dichos estudios no
han sufrido variación y, en caso de haber sufrido cambios,
estos se encuentren autorizados. El desarrollo del estudio
debe estar acorde con lo dispuesto en la Directiva vigente
al momento de solicitar la reinscripción.
Para la presentación de solicitudes de reinscripción
de las especialidades farmacéuticas que hayan
obtenido registro sanitario al amparo de la Ley N°
29459, incluyendo el requisito de sustento de seguridad
y eficacia correspondiente y cuya información de su
registro sanitario se encuentre actualizada, el titular
del registro sanitario quedará exceptuado de presentar
los requisitos señalados en los numerales 2, 3, 4,
9 y el inserto señalado en el numeral 8 del presente
literal. Asimismo, los estudios de estabilidad a largo
plazo, previstos en el numeral 6 de este artículo, se
tendrán por presentados en dicho procedimiento de
reinscripción, por medio de una declaración jurada que
exprese que dichos estudios no han sufrido variación y,
en caso de haber sufrido cambios, éstos se encuentren
autorizados. El desarrollo del estudio debe estar acorde
con lo dispuesto en la Directiva vigente al momento de
solicitar la reinscripción.
(...)".
"Artículo 100.- Condición de venta de los productos
dietéticos y edulcorantes
La condición de venta de los productos dietéticos
y edulcorantes es sin receta médica de venta en
establecimientos farmacéuticos y/o comerciales."
"Artículo 122.- Registro sanitario de los
dispositivos médicos
El registro sanitario de los dispositivos médicos se
otorga por nombre común, clasificación según nivel de
riesgo, fabricante y país del fabricante, adicionalmente
se otorgará por grupo de dispositivos (kit, set, sistema
y familia), nombre y país del sitio de fabricación,
nombre comercial y/o marca si los tuviera, tomando en
consideración los documentos de la International Medical
Devices Regulators Forum IMDRF.
Los datos anteriormente mencionados deben estar
avalados por el Certificado de Libre Comercialización.
Excepcionalmente, el nivel de riesgo, nombre común,
marca, familia, así como los componentes del kit o set que
no se encuentren detallados, pueden estar avalados por
carta del fabricante con el debido sustento.
En el caso que el fabricante declarado cuente con
distinto sitio de fabricación que elabore el dispositivo
médico, se debe informar además su nombre o razón
social y su dirección, el cual debe estar avalado por el
Certificado de Libre Comercialización.
En el caso que el fabricante declarado cuente con
distinto(s) sitio(s) de fabricación de accesorios, diferentes
al del dispositivo médico, se debe informar su razón social,
dirección y país de dicho(s) sitio(s), debiendo estos datos
estar incluidos en el Certificado de Libre Comercialización
o carta del fabricante.
Si de la combinación de dispositivos médicos resulta
un dispositivo que es destinado por el fabricante para
satisfacer un objetivo distinto al de los dispositivos
médicos individuales que lo componen, la combinación es
un nuevo dispositivo médico por derecho propio y deben
ser clasificados de acuerdo al nuevo uso previsto.
Si de la combinación de dispositivos médicos resulta
un dispositivo que es destinado para la comodidad del
usuario, pero no cambian los usos previstos individuales
de los mismos que la componen, la clasificación asignada
al conjunto de dispositivos corresponde a la del dispositivo
de más alto riesgo que se encuentra incluido en él.
El software que no viene incorporado en un dispositivo
médico, siempre que éste, de forma independiente se
encuentre enmarcado dentro de la definición de dispositivo
médico, debe clasificarse como sigue:
1. Cuando conduzca o ejerza influencia en el uso
particular del dispositivo médico, éste debe ser clasificado
de acuerdo al uso destinado de la combinación.
2. Cuando se encuentra de forma independiente de
cualquier otro dispositivo médico, se clasifica tomando en
consideración los documentos de la International Medical
Devices Regulators Forum - IMDRF.
3. El software de manera independiente (en la medida
que corresponda a la definición de un dispositivo médico)
será considerado como un dispositivo médico activo."
"Artículo 123.- Cambios en el registro sanitario de
dispositivos médicos
Los cambios de los dispositivos médicos con registro
sanitario, se clasifican en cambios de importancia
menor y cambios de importancia mayor, según su
nivel de riesgo para la salud de las personas o sus
repercusiones en la calidad, seguridad y eficacia del
dispositivo médico.
Para los cambios de importancia menor en el registro
sanitario, bastará la comunicación por escrito del titular
del registro sanitario a la Autoridad Nacional de Productos
Farmacéuticos, Dispositivos Médicos y Productos
Sanitarios (ANM) para que procedan automáticamente
dichos cambios, no siendo necesario que la citada
Autoridad emita pronunciamiento alguno. El titular del
registro sanitario tendrá un período de seis (6) meses
contados a partir del día siguiente de su comunicación
para implementar el(los) cambio(s) correspondiente(s).
Los cambios de importancia menor son especificados
en la Directiva aprobada por la Autoridad Nacional
de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarias.
Los cambios menores referidos a cambios
administrativos, como nombre comercial o razón social
y dirección del titular del registro sanitario, nombre del
director técnico u otros que defina la Autoridad Nacional
de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios (ANM), debidamente autorizados
por la Autoridad Nacional de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios (ANM)
correspondientes a un establecimiento farmacéutico como
titular del registro sanitario producirán automáticamente
sus efectos respecto a todos los rotulados, insertos y
manual de instrucciones de los dispositivos médicos de
los cuales sea titular de registro sanitario, sin necesidad
de efectuar trámite alguno.
Para los cambios de importancia mayor, el titular
del registro sanitario, dentro del período de su vigencia,
está obligado a presentar la solicitud de dicho cambio
ante la Autoridad Nacional de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios (ANM). La
Autoridad otorgará un período máximo de seis (6) meses
para la adecuación del cambio solicitado. Los cambios
de importancia mayor son especificados en la Directiva
aprobada por la Autoridad Nacional de Productos
Farmacéuticos, Dispositivos Médicos y Productos
Sanitarias.
Para la solicitud de cambios de importancia mayor
de la información declarada, se deben presentar los
siguientes documentos:
a)Solicitud con carácter de declaración jurada;
b) Documentos que sustenten el cambio, según
directiva específica;
Excepcionalmente, para los cambios de importancia
mayor, motivados por razones sanitarias que pudieran
afectar la salud pública, la Autoridad Nacional de
Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios (ANM), dispondrá que el titular
del registro sanitario implemente el referido cambio

561279
NORMAS LEGALES
Sábado 12 de setiembre de 2015
El Peruano
/
en un plazo específico, el cual será establecido por
la Resolución respectiva de la Autoridad Nacional de
Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios (ANM)."
"Artículo 138.- Requisitos que debe contener el
rotulado de los envases mediato e inmediato de los
dispositivos médicos
El proyecto de rotulado debe contener la siguiente
información:
a) Nombre del dispositivo médico;
b) Contenido del envase;
c) Si corresponde, la palabra "ESTERIL", indicando
método o simbología;
d) El código del lote o frase similar o el número de
serie, según corresponda, puede usar simbología;
e) Fecha de vencimiento o la indicación relacionada
a la fecha de vencimiento del dispositivo médico; cuando
no haya ninguna indicación relacionada a la fecha de
vencimiento, se debe consignar la fecha de fabricación,
según corresponda;
f) Finalidad de uso del dispositivo, no será necesaria
la finalidad de uso siempre que el dispositivo pueda ser
utilizado en forma correcta de acuerdo a su naturaleza;
g) Indicación de que el dispositivo contiene o incorpora
una sustancia medicinal o biológica, en caso corresponda;
h) Se acepta la frase de "un solo uso" o frase similar o
símbolo cuando corresponda, siempre y cuando éstos no
conlleven a confusión del usuario;
i) Las condiciones de almacenamiento, conservación
y/o manipulación del dispositivo médico, cuando
corresponda;
j) Las instrucciones especiales para operación y/o uso
de los dispositivos médicos, cuando así lo requiera;
k) Cualquier advertencia y/o precaución que deba
adoptarse;
l) Uso pediátrico, cuando corresponda;
m) Nombre y país del fabricante;
Se debe consignar el país del sitio de fabricación,
siempre y cuando éste sea diferente al país del fabricante:
1. En el caso de dispositivos fabricados en el
extranjero y envasados y acondicionados en el Perú, se
debe colocar: Símbolo o frase "Fabricado por....(nombre
y país del fabricante) ", y "envasado, acondicionado por
....(nombre del laboratorio nacional) para ......(titular
que registra el producto) ". Se acepta frase similar para
"fabricado por..."
2. Para dispositivos terminados fabricados en el
extranjero e importados al Perú, se debe colocar:
Símbolo o frase "Fabricado por.... (nombre y país del
fabricante) ", e "importado por... (titular que registra el
dispositivo) ". En caso de reacondicionamiento se debe
colocar adicionalmente reacondicionado por....(nombre
del laboratorio nacional) ". Se acepta frase similar para
"fabricado por..."
3. Cuando se trate de dispositivos terminados
fabricados en el país o en el extranjero por encargo de
un tercero se debe colocar: Símbolo o frase: "Fabricado
por... (nombre y país del fabricante) ", "para... (nombre de
la empresa que encarga su fabricación) ". Se acepta frase
similar para "fabricado por...".
n) Datos del titular de Registro Sanitario, en donde
se consigne el nombre, dirección y Registro Único de
Contribuyentes;
o) Nombre del director técnico;
p) Número de registro sanitario utilizando las siglas
"RS Nº..." o la frase: "Registro Sanitario Nº.......";
q) Número de lote: "Lote Nº......... " o la frase "Lote de
fabricación Nº........." o frase: similar o símbolo o número
de serie.
Para los dispositivos médicos que contengan
envase mediato e inmediato, en casos debidamente
sustentados y con carácter excepcional, los envases
inmediatos del dispositivo médico que, por su tamaño
pequeño, no pueden contener toda la información a que
se refiere el presente artículo, deben consignar, cuando
menos:
a) Nombre del dispositivo médico o código
debidamente sustentado;
b) Número de lote: "Lote Nº......... " o la frase: "Lote de
fabricación Nº........." o frase similar o símbolo o número
de serie;
c) Fecha de Vencimiento, cuando corresponda;
d) Condición de almacenamiento o simbología,
cuando corresponda;
e) Número de registro sanitario utilizando las siglas
"RS Nº..." o la frase: "Registro Sanitario Nº.......";
f) Cuando corresponda, la palabra «ESTÉRIL», frase
equivalente o símbolo.
En casos debidamente sustentados y con carácter
excepcional, los envases mediatos del dispositivo médico
y, para los dispositivos que sólo contienen envase
inmediato, que por su tamaño pequeño, no puedan
contener toda la información a que se refiere el presente
artículo, deben consignar, cuando menos:
a) Nombre del dispositivo médico;
b) Número de lote: "Lote Nº......... " o la frase: "Lote de
fabricación Nº........." o frase similar o símbolo o número
de serie;
c) Fecha de Vencimiento, cuando corresponda;
d) Condiciones especiales de almacenamiento y
transporte, cuando corresponda, o simbología;
e) Número de registro sanitario utilizando las siglas
"RS Nº..." o la frase: "Registro Sanitario Nº.......";
f) Finalidad de uso del dispositivo, si corresponde;
g) Simbología de seguridad y precauciones, y de los
cuidados especiales para el uso del dispositivo médico,
cuando corresponda;
h) Nombre del director técnico;
i) Datos del titular del registro sanitario, en donde
se consigne nombre, dirección y Registro Único de
Contribuyentes;
j) Nombre y país del fabricante y país del sitio de
fabricación;
k) Tratándose de dispositivos nacionales debe
consignarse adicionalmente el Registro Único de
Contribuyentes;
l) Cuando corresponda, la palabra «ESTÉRIL»,
frase equivalente o símbolo consignando el método o,
indicación de cualquier estado especial microbiológico o
de limpieza.
Para el caso de dispositivos médicos importados
estériles o no estériles con envase mediato sellado
completamente, cuya forma de presentación incluye
sólo una unidad, kit o set, debidamente sustentado por
el fabricante, el rotulado inmediato debe cumplir con lo
siguiente:
a) Nombre del dispositivo médico o código. En el caso
de kit o set nombre de cada componente o código, según
corresponda;
b) Número de lote: "Lote Nº........." o la frase: "Lote de
fabricación Nº........." o frase similar o símbolo o número
de serie;
c) Fecha de Vencimiento, cuando corresponda;
d) Condiciones especiales de almacenamiento y
transporte, cuando corresponda;
e) La denominación «ESTÉRIL», frase equivalente o
símbolo indicando el método, cuando corresponda;
f) Finalidad de uso del dispositivo, cuando corresponda;
g) Simbología de seguridad y precauciones, de los
cuidados especiales para el uso del dispositivo médico,
cuando corresponda;
h) Nombre y país del fabricante.
Cuando por las dimensiones pequeñas del envase
inmediato, el rotulado no pueda contener toda la
información antes mencionada, debe cumplir sólo con los
literales a), b), c), d) y e)."
"Artículo 174.- Plazo para presentar lo requerido
para el análisis de los productos o dispositivos
pesquisados
Para el análisis de las unidades de las muestras
pesquisadas de un producto o dispositivo, el titular
561280
NORMAS LEGALES
Sábado 12 de setiembre de 2015 /
El Peruano
del registro sanitario, de la notificación sanitaria
obligatoria o del certificado de registro sanitario debe
remitir, en un plazo máximo de treinta (30) días todos
los estándares (primarios y/o secundarios, internos,
de resolución, patrones de comparación y otros)
requeridos en la técnica analítica, contados a partir del
día siguiente de haber sido notificado por la Autoridad
Nacional de Productos Farmacéuticos, Dispositivos
Médicos y Productos Sanitarios, vencidos los cuales se
suspenderá el registro sanitario, notificación sanitaria
obligatoria o el certificado de registro sanitario del
producto o dispositivo por un periodo de ciento veinte
(120) días calendario, suspensión que puede ser
levantada si se remiten los requerimientos solicitados
y se cuenta con resultados de control de calidad
conformes. Transcurrido el plazo de ciento veinte (120)
días calendario, y de persistir en el incumplimiento
de esta disposición, se procede a cancelar el registro
sanitario, certificado de registro sanitario o notificación
sanitaria obligatoria del producto o dispositivo.
(...)
El certificado de análisis del producto terminado
correspondiente al lote pesquisado debe ser entregado
hasta cinco (5) días hábiles contados a partir del día
siguiente de haber sido notificado por la Autoridad Nacional
de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios, de acuerdo al formato respectivo
publicado en la página web de la citada Autoridad".
Artículo 2.- Modificación del Anexo N° 01 del
Reglamento para el Registro, Control y Vigilancia
Sanitaria de Productos Farmacéuticos, Dispositivos
Médicos y Productos Sanitarios
Modifíquese el numeral 41, e incorpórese los
numerales 110, 111, 112, 113, 114, 115, 116, 117 y 118
al Anexo N° 01 del Reglamento para el Registro, Control
y Vigilancia Sanitaria de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios aprobado
por Decreto Supremo Nº 016-2011-SA, de acuerdo al
siguiente detalle:
"ANEXO N° 01: GLOSARIO DE TERMINOS Y
dEFINICIONES
(...)
41.
Fabricante: Empresa que se encarga de todas
las operaciones que incluyen la adquisición de insumos
o componentes y productos, producción, empaque o
acondicionamiento, reacondicionamiento, aseguramiento
de la calidad y control de calidad, liberación,
almacenamiento y distribución de productos terminados y
los controles relacionados con estas operaciones.
Para el caso de dispositivos médicos se considera
fabricante a la persona natural o jurídica responsable
del diseño, fabricación, empaque o acondicionamiento,
ensamblado y rotulado de un dispositivo médico para
su comercialización. El fabricante será el responsable
del producto final, independientemente que las
operaciones antes mencionadas sean realizadas o no
por esta misma persona o en su nombre por otra(s)
persona(s).
(...)
110.
Estudios de estabilidad acelerados: Estudios
diseñados para lograr el incremento de la velocidad de
degradación química o física de un producto, mediante
condiciones de almacenamiento extremas o exageradas
en su envase original, con el propósito de monitorear las
reacciones de degradación y predecir el período de vida
bajo condiciones normales de almacenamiento.
111.
Estudios de estabilidad a largo plazo: Son
estudios diseñados de las características físicas, químicas
y microbiológicas, bajo condiciones de almacenamiento
controladas, durante el período de vida útil propuesto
del producto en el envase que se propone circular en el
mercado.
112.
Familia de dispositivos médicos: Conjunto
de dispositivos médicos que son utilizados para la
misma indicación de uso, poseen el mismo principio de
funcionamiento o mecanismo de acción, son elaborados
por el mismo fabricante y que cada producto que lo
constituye contiene características semejantes. Posee
las mismas precauciones, restricciones, advertencias,
cuidados especiales y aclaraciones sobre el uso del
dispositivo médico, como su almacenamiento y transporte.
Los dispositivos médicos estériles y no estériles no
pueden ser agrupados en una misma familia si tienen
diferente nivel de riesgo.
113.
Farmacovigilancia espontánea: Método
basado en la comunicación, recolección y evaluación
de notificaciones, realizadas por un profesional de la
salud, de sospechas de reacciones adversas a productos
farmacéuticos.
114.
Formato de Notificación de Sospecha de
Reacción Adversa: Conocido internacionalmente
como "Hoja amarilla". Es el formulario de recogida
de sospechas de reacciones adversas, aprobado por
la Autoridad Nacional de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios (ANM),
recoge información relativa al paciente (identificación,
edad, sexo, peso), al producto farmacéutico
sospechoso (nombre, dosis, frecuencia, fecha de inicio
y final, indicación terapéutica), a la reacción adversa
(descripción, fecha de comienzo y final, desenlace,
efecto de la reexposición si ha existido, entre otros)
y al profesional notificador (nombre, dirección/correo
electrónico, teléfono, profesión, entre otros).
115.
Kit: Conjunto de dispositivos médicos
complementarios que interactúan entre sí y que se
suministra como un todo, destinados a utilizarse en
la misma determinación o en el mismo procedimiento
médico.
116.
Set: Conjunto de dispositivos médicos con
características idénticas o similares, utilizados para un
mismo fin, y que se diferencian entre si únicamente en
color, tamaño o aroma y son comercializados como un
todo.
117.
Sistema de dispositivos médicos: Dispositivo
médico constituido por componentes complementarios
y compatibles de uso exclusivo entre sí, para una
función única y específica, que mantienen relación
de interdependencia para obtener una funcionalidad
destinada a efectuar un determinado procedimiento
médico y cuyo desempeño únicamente es obtenido si los
componentes son utilizados de forma integrada.
118.
Sitio de fabricación: Es la planta donde el
fabricante realiza sus actividades de fabricación, empaque
o acondicionamiento, ensamblado y etiquetado de un
dispositivo médico."
Artículo 3.- Modificación del Anexo 04 del
Reglamento para el Registro, Control y Vigilancia
Sanitaria de Productos Farmacéuticos, Dispositivos
Médicos y Productos Sanitarios
Modifíquese el sexto símbolo del Anexo 04 del
Reglamento para el Registro, Control y Vigilancia Sanitaria
de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios aprobado por Decreto Supremo Nº
016-2011-SA, de acuerdo al siguiente detalle:
y final, desenlace, efecto de la reexposición si ha existido, entre otros) y al
profesional notificador (nombre, dirección/correo electrónico, teléfono, profesión,
entre otros).
115.
Kit: Conjunto de dispositivos médicos complementarios que interactúan entre sí y
que se suministra como un todo, destinados a utilizarse en la misma
determinación o en el mismo procedimiento médico.
116.
Set: Conjunto de dispositivos médicos con características idénticas o similares,
utilizados para un mismo fin, y que se diferencian entre si únicamente en color,
tamaño o aroma y son comercializados como un todo.
117.
Sistema de dispositivos médicos: Dispositivo médico constituido por
componentes complementarios y compatibles de uso exclusivo entre sí, para una
función única y específica, que mantienen relación de interdependencia para
obtener una funcionalidad destinada a efectuar un determinado procedimiento
médico y cuyo desempeño únicamente es obtenido si los componentes son
utilizados de forma integrada.
118.
Sitio de fabricación: Es la planta donde el fabricante realiza sus actividades de
fabricación, empaque o acondicionamiento, ensamblado y etiquetado de un
dispositivo médico."
Artículo 3.- Modificación del Anexo 04 del Reglamento para el Registro,
Control y Vigilancia Sanitaria de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios
Modifíquese el sexto símbolo del Anexo 04 del Reglamento para el Registro,
Control y Vigilancia Sanitaria de Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios aprobado por Decreto Supremo Nº 016-2011-SA, de acuerdo al
siguiente detalle:
FABRICANTE
Artículo 4.- Modificación del Anexo 05 del
Reglamento para el Registro, Control y Vigilancia
Sanitaria de Productos Farmacéuticos, Dispositivos
Médicos y Productos Sanitarios
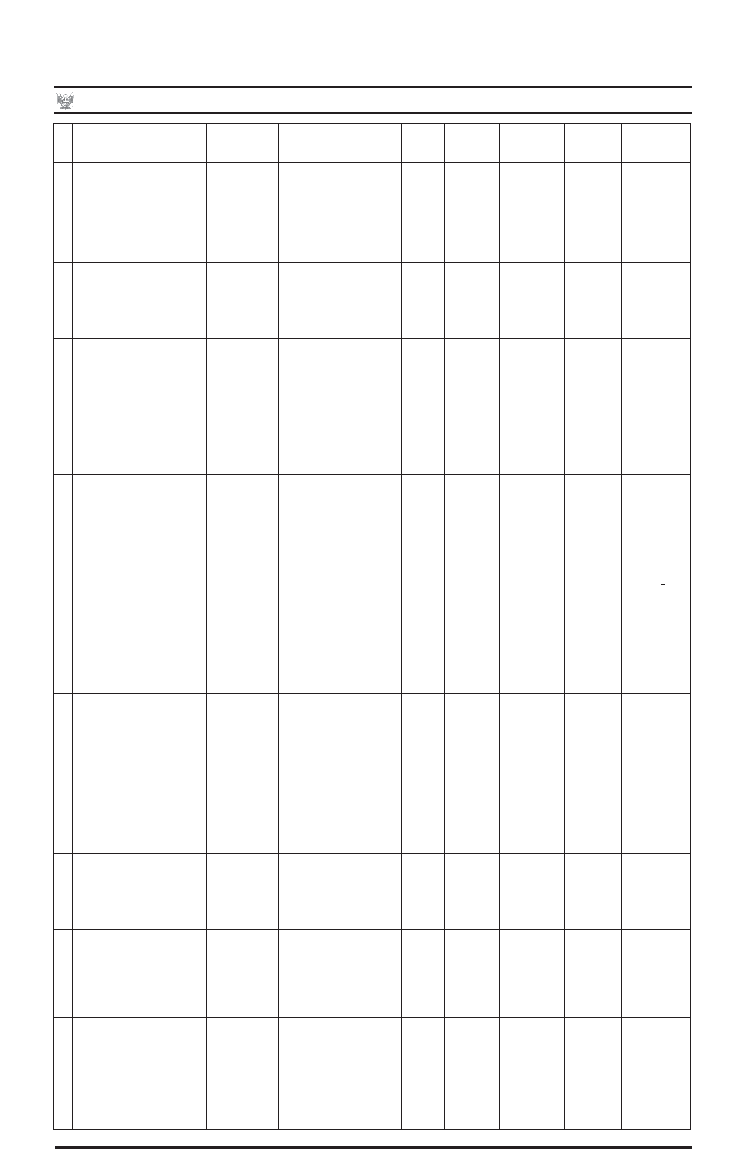
Modifíquense las infracciones 1, 2, 6, 7, 21, 41, 49 y
54 en el Anexo 05: Escala de Infracciones y Sanciones
Administrativas al Reglamento para el Registro, Control
y Vigilancia Sanitaria de Productos Farmacéuticos,
Dispositivos Médicos y Productos Sanitarios, aprobado
por Decreto Supremo Nº 016-2011-SA, de acuerdo al
siguiente detalle:
561281
NORMAS LEGALES
Sábado 12 de setiembre de 2015
El Peruano
/
INFRACCIÓN
FARMACIA
o BOTICA
FARMACIA DE LOS
ESTABLECIMIENTOS
DE SALUD
BOTIQUÍN DROGUERÍAS
ALMACÉN
ESPECIALIZADO LABORATORIO
NO
FARMACÉUTICO
1
Por no comunicar cambios de
importancia menor de productos
farmacéuticos o dispositivos
médicos.
Artículo 45 de la Ley N° 29459
Artículos 36 y 123 del Reglamento,
aprobado por D.S. N° 016-2011-
SA
NA
NA
NA
1 UIT
NA
1 UIT
NA
2
Por comercializar productos o
dispositivos sin haber solicitado los
cambios de importancia mayor en
su Registro Sanitario. Artículos 36
y 123 del Reglamento, aprobado
por D.S. N° 016-2011-SA
NA
NA
NA
3 UIT
NA
3 UIT
NA
6
Por fabricar, importar, almacenar,
distribuir, comercializar, expender o
dispensar productos o dispositivos
con registro sanitario, notificación
sanitaria obligatoria o certificado
de registro sanitario vencido,
suspendido o cancelado, excepto
para aquellos en que se autorizó el
agotamiento de stock Artículos 5, 7
y 13 del Reglamento, aprobado por
D.S. N° 016-2011-SA.
3 UIT
o
Cierre temporal
por 30 días
o
Cierre definitivo
3 UIT
o
Cierre temporal por 30 días
o
Cierre definitivo
0.5 UIT
o
Cierre
temporal
por 30
días
o
Cierre
definitivo
10 UIT
o
Cierre
temporal por
30 días
o
Cierre
definitivo
3 UIT
o
Cierre temporal
por 30 días
10 UIT
o
Cierre
temporal por
30 días
o
Cierre
definitivo
3 UIT
7
Por fabricar, importar, almacenar,
distribuir, comercializar o
dispensar productos o dispositivos
sin consignar o modificar en el
rotulado del envase mediato o
inmediato información técnica
aprobada en el registro sanitario
u otra información exigida en las
normas sanitarias de rotulado, o
consignando información técnica
no autorizada, con excepción
del número de lote, fecha
de expiración, y de aquellos
productos o dispositivos a los que
se les autorizó el agotamiento de
stock. Artículos 16, 17 y 18 del
Reglamento, aprobado por D.S.
N°016-2011-SA.
NA
NA
NA
3UIT
NA
3 UIT
NA
21
Por no remitir la cantidad
suficiente de los estándares
(primarios o secundarios, internos,
de resolución, patrones de
comparación y otros), necesarios
para realizar los ensayos
completos, de acuerdo a la técnica
analítica del producto pesquisado,
dentro del plazo establecido en
el artículo 174 del Reglamento,
aprobado por D.S. 016-2011-SA.
Artículo 175 del Reglamento,
aprobado por D.S. N° 016-2011-SA
NA
NA
NA
2 UIT
NA
2 UIT
NA
41
Por no cumplir el ensayo de
características físicas de polvos
estériles para reconstituir en
soluciones de inyectables. Artículo
5 del Reglamento, aprobado por
D.S. N° 016-2011-SA
NA
NA
NA
5 UIT
NA
5 UIT
NA
49
Por efectuar cambios en los
excipientes en la fórmula y/o
materiales de los envases
inmediato sin la autorización
correspondiente. Artículos 5 y 15
del Reglamento, aprobado por
D.S. N°016-2011-SA
NA
NA
NA
10 UIT
NA
10 UIT
NA
54
Por no actualizar las
especificaciones del producto o
dispositivo, con el que se registró
de acuerdo a la última edición de
la farmacopea o suplemento o
técnica propia o texto de referencia
cuando corresponda. Artículos 6 y
31 del Reglamento, aprobado por
D.S. N°016-2011-SA
NA
NA
NA
2 UIT
NA
2 UIT
NA

561282
NORMAS LEGALES
Sábado 12 de setiembre de 2015 /
El Peruano
Artículo 5.- Refrendo
El presente Decreto Supremo es refrendado por el
Ministro de Economía y Finanzas y el Ministro de Salud.
dISpOSICIÓN
COMpLEMENTARIA TRANSITORIA
Única.- Excepcionalmente, los titulares de registro
sanitario y de aquellas solicitudes que se encuentren
en trámite de registro sanitario de dispositivos médicos,
antes de la vigencia del presente Decreto Supremo,
podrán solicitar el reacondicionamiento del nombre del
sitio de fabricación, siempre y cuando en el rotulado figure
el nombre del fabricante.
dISpOSICIÓN
COMPLEMENTARIA DEROGATORIA
Única.- Deróguese el artículo 182 del Reglamento
para el Registro, Control y Vigilancia Sanitaria de
Productos Farmacéuticos, Dispositivos Médicos y
Productos Sanitarios, aprobado por Decreto Supremo Nº
016-2011-SA y la Resolución Ministerial N° 437-98-SA/
DM, que aprueba la Directiva de Pesquisa de Productos
Farmacéuticos y Afines.
Dado en la Casa de Gobierno, en Lima, a los once
días del mes de setiembre del año dos mil quince.
OLLANTA HUMALA TASSO
Presidente de la República
ALONSO SEGURA VASI
Ministro de Economía y Finanzas
ANÍBAL VELÁSQUEZ VALDIVIA
Ministro de Salud
1286856-6
Designan funcionarios en la Dirección
General de Salud de las Personas del
Ministerio
RESOLUCIÓN MINISTERIAL
Nº 563-2015/MINSA
Lima, 9 de setiembre del 2015
Vistos, los expedientes Nºs. 15-081026-001 y 15-
081655-001, que contienen las Notas Informativas Nºs.
948 y 949-2015-DGSP/MINSA, emitidas por la Directora
General de la Dirección General de Salud de las Personas
del Ministerio de Salud; y,
CONSIDERANDO:
Que, mediante Resolución Ministerial Nº 1019-2014/
MINSA, de fecha 31 de diciembre de 2014, se aprobó
el Cuadro para Asignación de Personal Provisional del
Ministerio de Salud, y mediante Resolución Ministerial
Nº 1030-2014/MINSA se aprobó la modificación del
citado instrumento de gestión, en el cual los cargos
de Ejecutivo/a Adjunto/a I y Director/a Ejecutivo/a
de la Dirección de Atención Integral de la Salud, de
la Dirección General de Salud de las Personas, se
encuentran calificados como Directivo Superior de
Libre Designación y de confianza, respectivamente;
Que, por Resolución Ministerial Nº 982-2014/
MINSA, de fecha 17 de diciembre de 2014, se
designó a la médico cirujano Marina Antonieta Ochoa
Linares, en el cargo de Directora Ejecutiva, Nivel F-4,
de la Dirección de Atención Integral de la Salud de
la Dirección General de Salud de las Personas del
Ministerio de Salud;
Que, según Resolución Ministerial Nº 035-2015/
MINSA, de fecha 19 de enero de 2015, se designó al
médico cirujano Víctor Javier Suárez Moreno, en el
cargo de Ejecutivo Adjunto I, Nivel F-4, de la Dirección
General de Salud de las Personas del Ministerio de
Salud;
Que, mediante Nota Informativa Nº 948-2015-DGSP/
MINSA, la Directora General de la Dirección General de
Salud de las Personas comunica de la renuncia formulada
por el médico cirujano Víctor Javier Suárez Moreno, al
cargo en el que fuera designado mediante Resolución
Ministerial Nº 035-2015/MINSA, y propone designar en su
reemplazo a la médico cirujano Marina Antonieta Ochoa
Linares;
Que, asimismo, con Nota Informativa Nº 949-2015-
DGSP/MINSA, solicita dar por concluida la designación
de la médico cirujano Marina Antonieta Ochoa Linares,
en el cargo de Directora Ejecutiva de la Dirección de
Atención Integral de la Salud y propone designar en su
reemplazo a la médico cirujano Zila Patricia Caballero
Ñopo;
Que, a través del Informe Nº 698-2015-EIE-
OARH/MINSA, remitido mediante Memorando Nº
1668-2015-OGGRH-OARH-EIE/MINSA,
la
Oficina
General de Gestión de Recursos Humanos, emite opinión
favorable en relación a lo solicitado por la Directora
General de la Dirección General de Salud de las Personas,
señalando que procede adoptar las acciones de personal
propuestas a fin de asegurar el normal funcionamiento de
la citada Dirección General;
Con el visado de la Directora General de la Oficina
General de Gestión de Recursos Humanos, de la Directora
General de la Oficina General de Asesoría Jurídica, del
Viceministro de Salud Pública y de la Secretaria General;
y,
De conformidad con lo previsto en la Ley Nº 27594,
Ley que regula la participación del Poder Ejecutivo
en el nombramiento y designación de funcionarios
públicos; en la Ley Nº 29849, Ley que establece la
eliminación progresiva del Régimen Especial del
Decreto Legislativo 1057 y otorga derechos laborales;
en el Reglamento del Decreto Legislativo Nº 1057,
que regula el Régimen Especial de Contratación
Administrativa de Servicios, aprobado por Decreto
Supremo Nº 075-2008-PCM y sus modificatorias;
en el numeral 8) del artículo 25 de la Ley Nº 29158,
Ley Orgánica del Poder Ejecutivo y en el Decreto
Legislativo Nº 1161, Ley de Organización y Funciones
del Ministerio de Salud;
SE RESUELVE:
Artículo 1.- Dar por concluida la designación de la
médico cirujano Marina Antonieta Ochoa Linares, en el
cargo de Directora Ejecutiva, Nivel F-4, de la Dirección
de Atención Integral de la Salud de la Dirección General
de Salud de las Personas del Ministerio de Salud,
dándosele las gracias por los servicios prestados.
Artículo 2.- Aceptar la renuncia del médico cirujano
Víctor Javier Suárez Moreno, al cargo de Ejecutivo
Adjunto I, Nivel F-4, de la Dirección General de Salud
de las Personas del Ministerio de Salud, dándosele las
gracias por los servicios prestados.
Artículo 3.- Designar en la Dirección General de
Salud de las Personas del Ministerio de Salud, a las
profesionales que se detallan a continuación:
NOMBRES Y APELLIDOS
CARGO
NIVEL
Médico cirujano Zila Patricia
Caballero Ñopo
Directora Ejecutiva de
la Dirección de Atención
Integral de la Salud
F-4
Médico cirujano Marina
Antonieta Ochoa Linares
Ejecutiva Adjunta I
F-4
Regístrese, comuníquese y publíquese.
ANÍBAL VELÁSQUEZ VALDIVIA
Ministro de Salud
1285909-1